
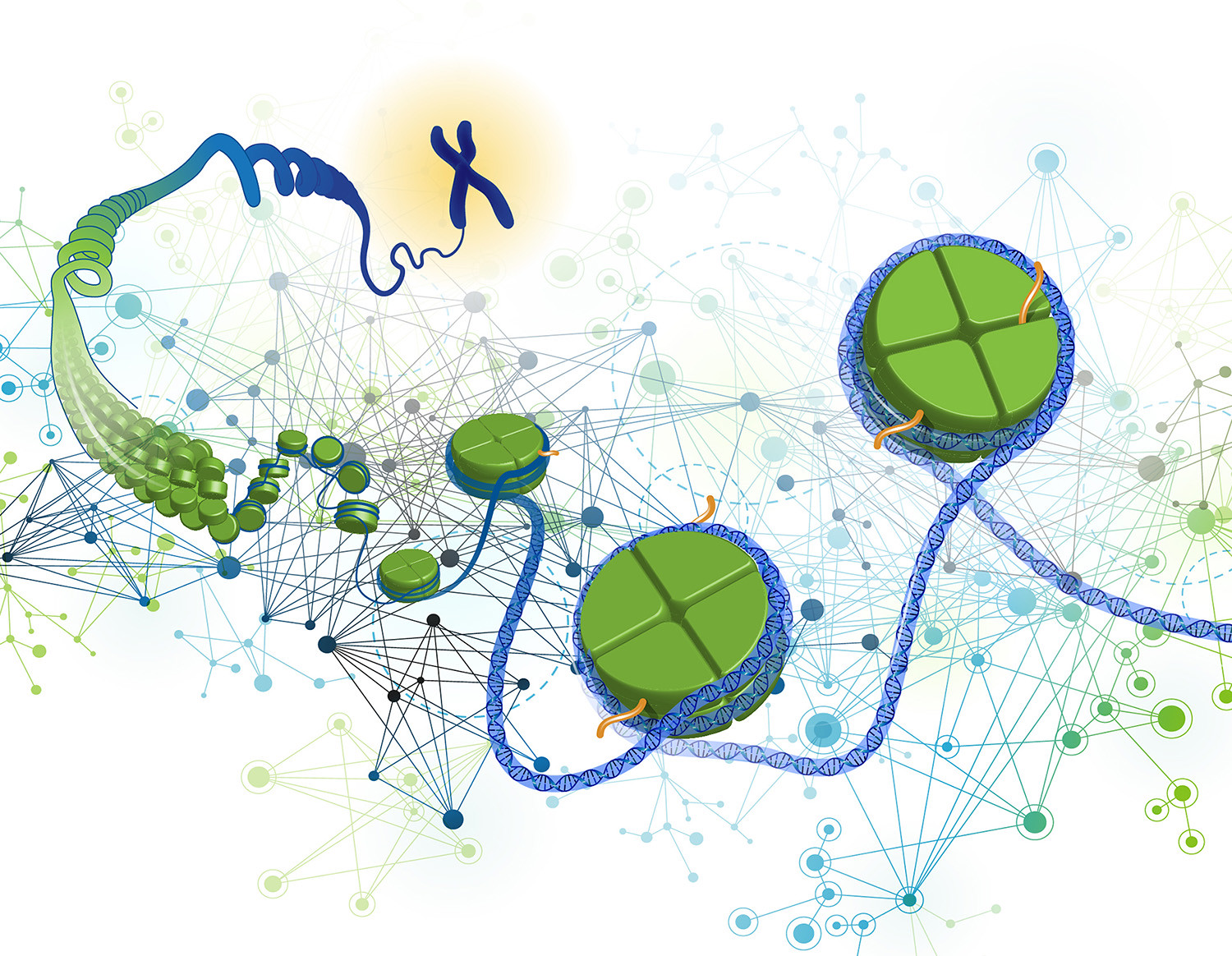
There is a fundamental problem at the heart of all cancers: cells that won’t stop dividing, or hyperproliferation, along with genomic instability. This runaway growth is what forms a tumor, and the abnormal cellular processes that drive this growth also thwarts therapies that target cancer cells.
Despite more than six decades of research into the mechanisms that cells use to divide, some of the nuts and bolts of the process remain a mystery. Scientists want to better understand these mechanisms in hopes of targeting them and potentially shutting down the uncontrolled growth of some tumors.
New research from three collaborating teams of scientists in the United States and Europe appears to have found one of these mechanisms, uncovering a previously unknown check on dividing cells: a protein called AMBRA1. By limiting tumor growth, the researchers showed, AMBRA1 serves as an important tumor suppressor—as these types of molecules are often called—in healthy cells.
The new work shows that AMBRA1 marks other proteins involved in helping cells divide (known as cyclins) for destruction when cell division isn’t needed. When the gene that produces AMBRA1 is damaged and the protein is missing or doesn’t work properly, the cell cycle loses one of its main brakes, potentially letting cell division spiral out of control.
The loss of the AMBRA1 protein, the researchers found, can drive tumor formation in mice and is linked with worse outcomes in some human tumors. They also reported that a lack of AMBRA1 may make some tumors resistant to drugs called CDK4/6 inhibitors, which are a promising new class of cancer treatments. Another axis of therapy development involves synthetic lethality.
The new research may point to potential strategies for blocking the runaway cell division caused by loss of AMBRA1 and for re-sensitizing cancer cells to CDK4/6 inhibitors. The results from all three studies were published on April 14 in Nature.
“These findings may eventually lead to new treatments,” including those designed to combat resistance to CDK4/6 inhibition, said Stefan Maas, Ph.D., of NCI’s Division of Cancer Biology, who was not involved with the studies.
Uncovering AMBRA1
Although the respective research teams behind each study began their work on AMBRA1 separately, it soon became a collaborative effort.
Michele Pagano, M.D., of NYU Langone Health, and a student in his lab, Daniele Simoneschi, Ph.D., were working on writing up the results of their study for submission to a journal. Then, at a conference, they came across a similar study being presented by Andrea Chaikovsky, a doctoral student in the laboratory of Julien Sage, Ph.D., of Stanford Medicine. The two groups agreed to stay in touch and share information as their research progressed, Dr. Pagano said.
When Dr. Pagano’s team needed a mouse model to study the impact of alterations in the AMBRA1 gene, they contacted researchers at the Danish Cancer Society Research Center in Copenhagen, Denmark, who had developed such a model. When that group heard what the NYU and Stanford teams had initially found in their respective studies, they joined the effort, expanding their work to look at the effect of certain drugs in cancer cells missing the AMBRA1 protein.
“It was very interesting to see how we were able to both come together in a very collaborative and collegial spirit and, at the same time, keep all the studies very independent. The three papers were very different in how we approached the project,” said Dr. Sage.
Both US teams, which were funded in part by NCI, were looking for molecules that regulate D-type cyclins. When D-type cyclins build up in a cell, they bind to and activate the CDK4 and CDK6 proteins, spurring a complex set of interactions that eventually nudge cells into dividing. At that point, the levels of D-type cyclins drop, only to rise again when the cell gets a signal to divide.
When Dr. Pagano and his team screened cells for proteins that regulate the levels of D-type cyclins, they identified AMBRA1 with every method use
d. Next, they blocked the activity of AMBRA1 in cells and found that this caused D-type cyclins to build up in cells when they weren’t supposed to. Further experiments showed that AMBRA1’s role was to tag unneeded cyclins for destruction, thereby keeping cells from dividing out of turn.
The discovery that AMBRA1 helps control cyclins, and therefore cell division, raised the question of whether AMBRA1 is involved in cancer. Using data collected by The Cancer Genome Atlas (TCGA), the researchers found that low levels of AMBRA1 were a marker of poor outcomes for patients with several cancer types. And when they implanted human lymphoma cells that lacked AMBRA1 into mice, the resulting tumors grew much larger and faster than those derived from cells with intact AMBRA1.
In a final set of experiments, the team found that loss of AMBRA1 changed how cancer cells responded to a Food and Drug Administration (FDA)-approved CDK4/6 inhibitor called palbociclib (Ibrance). It turned out that the loss of AMBRA1 triggered D-type cyclins to bind to and activate another cell cycle protein, called CDK2, in addition to CDK4/6. By joining together with CDK2, the cyclins could bypass the effects of CDK4/6 inhibitors and continue to drive tumor growth.
A Problem and an Opportunity
The second study, from Dr. Sage’s team, found similar results. After identifying AMBRA1 as a regulator of D-cyclin levels in cells and finding that its loss blunts the effectiveness of CDK4/6 inhibitors, the team went on to look at its significance in lung cancer.
The team generated human lung cancer cells that lacked specific tumor suppressors and then implanted these cells into mice. Cells lacking AMBRA1 grew faster than those missing other tumor suppressor proteins tested, including one called RB, which is a commonly mutated tumor suppressor in the cell cycle pathway.
In the TCGA database, people with lung cancer whose tumors lacked AMBRA1 did not live as long as those with tumors that retained AMBRA1. However, the negative impact of AMBRA1 loss was limited to lung cancer patients who also had mutations in a gene called KRAS.
Further work is needed to understand the intricacies of these sorts of interactions with AMBRA1, but “there [can be] different wiring in cancer cells that make a tumor suppressor more prominent in one cancer versus another cancer,” Dr. Sage explained.
The third study, from the Danish team and their collaborators in Italy, focused on the consequences of AMBRA1 loss in cells. They found that the absence of AMBRA1 leads to a phenomenon called replication stress, which alters the DNA copying process that occurs when cells divide.
But the replication stress caused by AMBRA1 loss also presents an opportunity, their findings suggested. In other experiments in cells without AMBRA1, when the team blocked a protein called CHK1 that helps inhibit cell division in the presence of replication stress, the cells couldn’t stop dividing and died.
The findings suggest that loss of AMBRA1 may be, instead of a disaster, “a vulnerability that could potentially be exploited in cancer treatments,” the study authors wrote.
Targeting AMBRA1’s Absence
“Collectively, these studies indicate that AMBRA1 normally restrains cell proliferation, largely by stopping D-cyclins from reaching high levels,” wrote Charupong Saengboonmee, M.D., and Piotr Sicinski, M.D., Ph.D., of the Dana-Farber Cancer Institute, in an accompanying editorial.
“Taken together, they provide particularly robust results, since three different research groups using various approaches provide strong validation for the core findings,” said Dr. Maas. “AMBRA1 is established as a tumor suppressor that is mutated in human cancer.”
The clinical implications of the studies need to be teased out in future research, Dr. Sage explained. “AMBRA1 could potentially be used as a biomarker to determine whether a patient will respond to CDK4/6 inhibitors; that’s definitely worth looking at,” said Dr. Sage.
FDA has approved three CDK4/6 inhibitors, all for slightly different uses in people with metastatic breast cancer. They’re also being tested in clinical trials for many other cancer types. Having a biomarker to predict response could potentially spare some patients from unnecessary costs and side effects of CDK4/6 inhibitors, in cases where they’re unlikely to be effective.
Future work is also needed to test strategies for reversing resistance to CDK4/6 inhibitors driven by loss of AMBRA1. “A particular exciting possibility … is that CHK1 inhibitors can be used to treat CDK4/6-inhibitor-resistant tumors that have low levels of AMBRA1,” wrote Drs. Saengboonmee and Sicinski.
“Another possible way to target tumors that lack AMBRA1 may be shutting down CDK2, the molecule that helps shield cyclins from CDK4/6 inhibition in tumors with low levels of AMBRA1,” Dr. Pagano said. “Drugs that specifically target this molecule are currently in development by pharmaceutical companies.”
And there may be other ways to exploit AMBRA1’s role as a tumor suppressor, Dr. Sage said. “It’s often the case in basic science that you discover something, but the therapeutic implication is not obvious. But maybe 1, 2, 5, or 10 years later—then you realize why it was important.”
Source: Adapted from cancer.gov