
Proteins are the workhorses of the cell. Mapping the precise shapes of the most important of these workhorses helps to unlock their life-supporting functions or, in the case of disease, potential for dysfunction. While the amino acid sequence of a protein provides the basis for its 3D structure, deducing the atom-by-atom map from principles of quantum mechanics has been beyond the ability of computer programs—now, researchers can just “google” it…
In a recent study in the journal Science, researchers reported they have developed artificial intelligence approaches for predicting the three-dimensional structure of proteins in record time, based solely on their one-dimensional amino acid sequences.
This new National Institutes of Health (NIH)-supported advance is now freely available to scientists around the world. In fact, it has already helped to solve especially challenging protein structures in cases where experimental data were lacking and other modeling methods hadn’t been enough to get a final answer. It also can now provide key structural information about proteins for which more time-consuming and costly imaging data are not yet available.
The new work comes from a group led by David Baker and Minkyung Baek, University of Washington, Seattle, Institute for Protein Design. Over the course of the pandemic, Baker’s team has been working hard to design promising COVID-19 therapeutics. They’ve also been working to design proteins that might offer promising new ways to treat cancer and other conditions. As part of this effort, they’ve developed new computational approaches for determining precisely how a chain of amino acids, which are the building blocks of proteins, will fold up in space to form a finished protein.
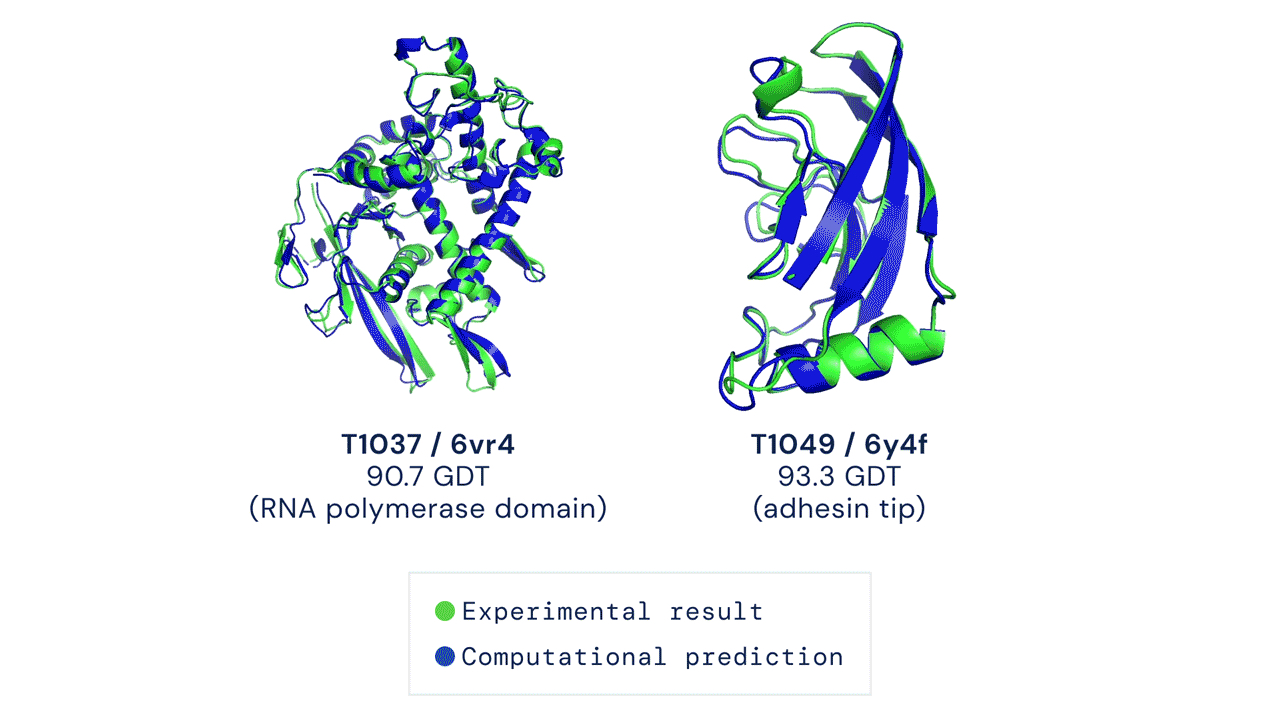
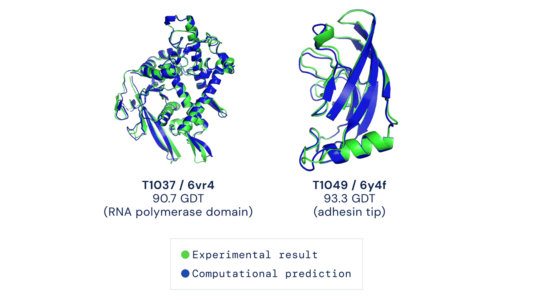
But the ability to predict a protein’s precise structure or shape from its sequence alone had proven to be a difficult problem to solve despite decades of effort. In search of a solution, research teams from around the world have come together every two years since 1994 at the Critical Assessment of Structure Prediction (CASP) meetings. At these gatherings, teams compete against each other with the goal of developing computational methods and software capable of predicting any of nature’s 200 million or more protein structures from sequences alone with the greatest accuracy.
DeepMind — a British AI research lab owned by U.S. technology giant Google — has created machine learning software called AlphaFold that solves a huge problem in biology: predicting the 3-D structure of a protein based on its sequence. Last week, it open-sourced its software and shared with the world a database of 350,000 predicted structures for the most useful proteins in science, including all human proteins. In fact, it could predict most protein structures almost as accurately as other high-resolution protein mapping techniques, including today’s go-to strategies of X-ray crystallography and cryo-EM.
The DeepMind performance showed what was possible, but because the advances were made by a world-leading deep learning company, the details on how it worked weren’t made publicly available at the time. The findings left Baker, Baek, and others eager to learn more and to see if they could replicate the impressive predictive ability of AlphaFold outside of such a well-resourced company.
In the new work, Baker and Baek’s team has made stunning progress—using only a fraction of the computational processing power and time required by AlphaFold. The new software, called RoseTTAFold, also relies on a deep learning approach. In deep learning, computers look for patterns in large collections of data. As they begin to recognize complex relationships, some connections in the network are strengthened while others are weakened. The finished network is typically composed of multiple information-processing layers, which operate on the data to return a result—in this case, a protein structure.
Biofacturing companies like Zymergen use these microbes to make products with biology. The DeepMind data on them is expected to help create microbes that better produce breakthrough chemicals and materials. “The DeepMind library could help the industry take a big step forward,” says Zymergen CTO Aaron Kimball, “whether that’s to make new biosynthetic pathways for chemicals and materials, or to make novel proteins for human therapeutic applications.”
Given the complexity of the problem, instead of using a single neural network, RoseTTAFold relies on three. The three-track neural network integrates and simultaneously processes one-dimensional protein sequence information, two-dimensional information about the distance between amino acids, and three-dimensional atomic structure all at once. Information from these separate tracks flows back and forth to generate accurate models of proteins rapidly from sequence information alone, including structures in complex with other proteins.
As soon as the researchers had what they thought was a reasonable working approach to solve protein structures, they began sharing it with their structural biologist colleagues. In many cases, it became immediately clear that RoseTTAFold worked remarkably well. What’s more, it has been put to work to solve challenging structural biology problems that had vexed scientists for many years with earlier methods.
RoseTTAFold already has solved hundreds of new protein structures, many of which represent poorly understood human proteins. The 3D rendering of a complex showing a human protein called interleukin-12 in complex with its receptor (above image) is just one example. The researchers have generated other structures directly relevant to human health, including some that are related to lipid metabolism, inflammatory conditions, and cancer. The program is now available on the web and has been downloaded by dozens of research teams around the world.
Cryo-EM and other experimental mapping methods will remain essential to solve protein structures in the lab. But with the artificial intelligence advances demonstrated by RoseTTAFold and AlphaFold, researchers now can make the critical protein structure predictions at their desktops. This newfound ability will be a boon to basic science studies and has great potential to speed life-saving therapeutic advances even for companies that do not normally employ machine learning technologies.
Source: Adopted from NIH.gov