
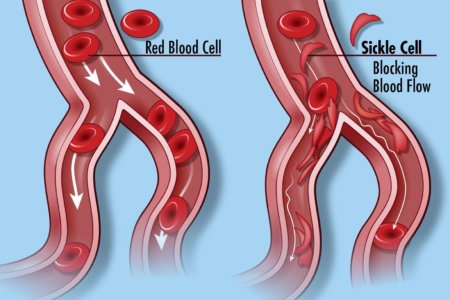
Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round, and they move through small blood vessels to carry oxygen to all parts of the body. In someone who has SCD, the red blood cells become hard and sticky and look like a farm tool called a “sickle.” The sickle cells die early, which causes a constant shortage of red blood cells. When they travel through small blood vessels, sickle cells get stuck and clog the blood flow. This can cause pain and other serious problems such infection, acute chest syndrome and stroke.
SCD is caused by a specific point mutation in a gene that codes for the beta chain of hemoglobin. People with just one copy of this mutation have sickle cell trait and are generally healthy. But those who inherit two mutant copies of this gene suffer lifelong consequences of the presence of this abnormal protein. Their red blood cells—normally flexible and donut-shaped—assume the sickled shape that gives SCD its name. The sickled cells clump together and stick in small blood vessels, resulting in severe pain, anemia, stroke, pulmonary hypertension, organ failure, and far too often, early death.

Recently, CBS’s “60 Minutes” profiled the story of Jennelle Stephenson , a young woman with SCD. Jennelle now appears potentially cured of this condition, thanks to an experimental gene therapy that was tested at the NIH Clinical Center in Bethesda, MD. As groundbreaking as this research may be, it’s among a variety of innovative strategies now being tried to cure SCD and other genetic diseases that have seemed out of reach.
Dr. Francis Collins was interviewed by 60 Minutes. He is director of the National Institutes of Health, the largest biomedical research agency in the world. He oversees a nearly 40 billion dollar budget that funds more than 400,000 researchers world-wide.
Dr. Collins was head of the Human Genome Project at the NIH in 2000 when he made a landmark announcement: after a decade of work, scientists had finally decoded the genes that make up a human being.


One exciting approach involves using gene editing to increase levels of fetal hemoglobin (HbF) in the red blood cells of people with SCD. Shortly after birth, babies usually stop producing HbF, and switch over to the adult form of hemoglobin. But rare individuals continue to make high levels of HbF throughout their lives. This is referred to as hereditary persistence of fetal hemoglobin (HPFH).
Individuals with HPFH are entirely healthy. Strikingly, rare individuals with SCD who also have HPFH have an extremely mild version of sickle cell disease—essentially the presence of significant quantities of HbF provides protection against sickling. So, researchers have been exploring ways to boost HbF in everyone with SCD—and gene editing may provide an effective, long-lasting way to do this.
Clinical trials of this approach are already underway. And new findings reported in Nature Medicine show it may be possible to make the desired edits even more efficiently, raising the possibility that a single infusion of gene-edited cells might be able to cure SCD.
Eleven years ago, a team led by Vijay Sankaran and Stuart Orkin at Boston Children’s Hospital and the Dana-Farber Cancer Institute discovered that a protein called BCL11A seemed to determine HbF levels. Subsequent work showed the protein actually works as a master mediator of the switch from fetal to adult hemoglobin, which normally occurs shortly after birth.
Five years ago, Orkin and Daniel Bauer identified a specific enhancer of BCL11A expression that could be an attractive target for gene editing. They could knock out the enhancer in the bone marrow, and BCL11A would not be produced, allowing HbF to stay switched on.
The researchers had another idea about turning off production of HbF in red cells. They thought it might be possible to keep HbF on permanently by disrupting BCL11A in blood-forming hematopoietic stem cells (HSCs). The hope was that such a treatment might offer people with SCD a permanent supply of healthy red blood cells.
Fast-forward to the present, and researchers are now testing the ability of gene editing tools to cure the disease.
CRISPR is a highly precise gene-editing tool that relies on guide RNA molecules to direct a scissor-like Cas9 enzyme to just the right spot in the genome to correct the misspelling. The gene-editing treatment involves removing bone marrow from a patient, modifying the HSCs outside the body using CRISPR gene-editing tools, and then returning them back to the patient. Preclinical studies had shown that CRISPR can be effective in editing BCL11A to boost HbF production.
But questions lingered about the editing efficiency in HSCs versus more common, shorter-lived progenitor cells found in bone marrow samples. The efficiency greatly influences how long the edited cells might benefit patients. Bauer’s team saw room for improvement and, as the new study shows, they were right.
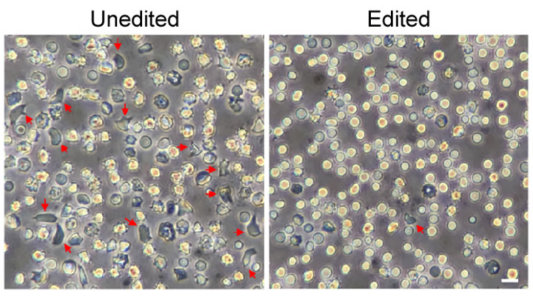
To produce lasting HbF production, it’s important to edit as many HSCs as possible. But it turns out that HSCs are more resistant to editing than other types of cells in bone marrow. With a series of adjustments to the gene-editing protocol, including use of an optimized version of the Cas9 protein, the researchers showed they could push the number of edited genes from about 80 percent to about 95 percent.
Their studies show that the most frequent Cas9 edits in HSCs are tiny insertions of a single DNA “letter.” With that slight edit to the BCL11A gene, HSCs reprogram themselves in a way that ensures long-term HbF production.
As a first test of their CRISPR-edited human HSCs, the researchers carried out the editing on HSCs derived from patients with SCD. Then they transferred the editing cells into immune-compromised mice. Four months later, the mice continued to produce red blood cells that produced high levels of HbF and resisted sickling. Bauer says they’re already taking steps to begin testing cells edited with their optimized protocol in a clinical trial.
What’s exciting is that the first U.S. human clinical trials of such a gene-editing approach for SCD are already underway, led by CRISPR Therapeutics/Vertex Pharmaceuticals and Sangamo Therapeutics/Sanofi. In January 2019, CRISPR Therapeutics/Vertex Pharmaceuticals announced that the U.S. Food and Drug Administration (FDA) had granted Fast Track Designation for their CRISPR-based treatment called CTX001.
Links:
Sickle Cell Disease (National Heart, Lung, and Blood Institute/NIH)
Cure Sickle Cell Initiative (NHLBI)
What are Genome Editing and CRISPR-Cas9? (National Library of Medicine/NIH)
Could Gene Therapy Cure Sickle Cell Anemia ? (CBS News)
Daniel Bauer (Dana-Farber Cancer Institute, Boston)
Somatic Cell Genome Editing Program (Common Fund/NIH)